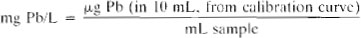
In order to promote public education and public safety, equal justice for all, a better informed citizenry, the rule of law, world trade and world peace, this legal document is hereby made available on a noncommercial basis, as it is the right of all humans to know and speak the laws that govern them.
18TH EDITION 1992
Prepared and published jointly by:
AMERICAN PUBLIC HEALTH ASSOCIATION
AMERICAN WATER WORKS ASSOCIATION
WATER ENVIRONMENT FEDERATION
Joint Editorial Board
Arnold E. Greenberg, APHA, Chairman
Lenore S. Clesceri, WEF
Andrew D. Eaton, AWWA
Managing Editor
Mary Ann H. Franson
Publication Office
American Public Health Association
1015 Fifteenth Street, NW
Washington, DC 20005
Copyright © 1917, 1920, 1923, and 1925 by
American Public Health Association
Copyright © 1933, 1936, and 1946 by
American Public Health Association
American Water Works Association
Copyright © 1955, 1960, and 1965 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1971 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1976 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1981 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1985 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1989 by
American Public Health Association
American Water Works Association
Water Pollution Control Federation
Copyright © 1992 by
American Public Health Association
American Water Works Association
Water Environment Federation
All rights reserved. No part of this publication may be reproduced, graphically or electronically, including entering in storage or retrieval systems, without the prior written permission of the publishers.
30M7/92
The Library of Congress has catalogued this work as follows:
American Public Health Association.
Standard methods for the examination of water and wastewater.
ISBN 0-87553-207-1
Printed and bound in the United States of America.
Composition: EPS Group, Inc., Hanover, Maryland
Set in: Times Roman
Printing: Victor Graphics, Inc., Baltimore, Maryland
Binding: American Trade Bindery, Baltimore, Maryland
Cover Design: DR Pollard and Associates, Inc., Arlington, Virginia
Lead is a serious cumulative body poison. Natural waters seldom contain more than 5 μg/L, although much higher values have been reported. Lead in a water supply may come from industrial, mine, and smelter discharges or from the dissolution of old lead plumbing. Tap waters that are soft, acid, and not suitably treated may contain lead resulting from an attack on lead service pipes or solder pipe joints.
The atomic absorption spectrometric method has a relatively high detection limit in the flame mode and requires an extraction procedure for the low concentrations common in potable water: the electrothermal atomic absorption method is much more sensitive for low concentrations and does not require extraction. The inductively coupled plasma method has a sensitivity similar to that of the flame atomic absorption method. The dithizone method is sensitive and specific as a colorimetric procedure.
*Approved by Standard Methods Committee. 1990.
See flame atomic absorption spectrometric method, Sections 3111B and C, and electrothermal atomic absorption spectrometric method, Section 3113.
See Section 3120.
a. Principle: An acidified sample containing microgram quantities of lead is mixed with ammoniacal citrate-cyanide reducing solution and extracted with dithizone in chloroform (CHCl3) to form a cherry-red lead dithizonate. The color of the mixed color solution is measured photometrically.1,2 Sample volume taken for analysis may be 2 L when digestion is used.
b. Interference: In a weakly ammoniacal cyanide solution (pH 8.5 to 9.5) dithizone forms colored complexes with bismuth, stannous tin, and monovalent thallium. In strongly ammoniacal citrate-cyanide solution (pH 10 to 11.5) the dithizonates of these ions are unstable and are extracted only partially.3 This method uses a high pH, mixed color, single dithizone extraction. Interference from stannous tin and monovalent thallium is reduced further when these ions are oxidized during preliminary digestion. A modification of the method allows detection and elimination of bismuth interference. Excessive quantities of bismuth, thallium, and tin may be removed.4
Dithizone in CHCl3 absorbs at 510 nm; control its interference by using nearly equal concentrations of excess dithizone in samples, standards, and blank.
The method is without interference for the determination of 0.0 to 30.0 μg Pb in the presence of 20 μg T1+, 100 μg Sn2+, 200 μg In3+, and 1000 μg each of Ba2+, Cd2+, Co2+, Cu2+, Mg2+, Mn2+, Hg2+, Sr2+, Zn2+, Al3+, Sb3+, As3+, Cr3+, Fe3+, V3+, PO33—, and SO42—. Gram quantities of alkali metals do not interfere. A modification is provided to avoid interference from excessive quantities of bismuth or tin.
c. Preliminary sample treatment: At time of collection acidify with conc HNO3 to pH <2 but avoid excess HNO3. Add 5 mL 0.1N iodine solution to avoid losses of volatile organo-lead compounds during handling and digesting of samples. Prepare a blank of lead-free distilled water and carry through the procedure.
d. Digestion of samples: Unless digestion is shown to be unnecessary, digest all samples for dissolved or total lead as described in 3030G or H.
e. Minimum detectable concentration: 1.0 μg Pb/10 mL dithizone solution.
a. Spectrophotometer for use at 510 nm, providing a light path of 1 cm or longer.
b. pH meter.
3-69c. Separatory funnels: 250-mL Squibb type. Clean all glassware, including sample bottles, with 1 + 1 HNO3. Rinse thoroughly with distilled or deionized water.
d. Automatic dispensing burets: Use for all reagents to minimize indeterminate contamination errors.
Prepare all reagents in lead-free distilled water.
a. Stock lead solution: Dissolve 0.1599 g lead nitrate. Pb(NO3)2 (minimum purity 99.5%), in approximately 200 mL water. Add 10 mL conc HNO3 and dilute to 1000 mL with water. Alternatively, dissolve 0.1000 g pure Pb metal in 20 mL 1 + 1 HNO3 and dilute to 1000 mL with water; 1.00 mL = 100 μg Pb.
b. Working lead solution: Dilute 2.0 mL stock solution to 100 mL with water; 1 mL = 2.00 μg Pb.
c. Nitric acid, HNO3, 1 + 4: Dilute 200 mL conc HNO3 to 1 L with water.
d. Ammonium hydroxide, NH4OH, 1 + 9: Dilute 10 mL conc NH4OH to 100 mL with water.
e. Citrate-cyanide reducing solution: Dissolve 400 g dibasic ammonium citrate, (NH4)2HC6H5O7, 20g anhydrous sodium sulfite, Na2SO3, 10 g hydroxylamine hydrochloride, NH2OH·HCl, and 40 g potassium cyanide, KCN (CAUTION: Poison) in water and dilute to 1 L. Mix this solution with 2 L conc NH4OH. Do not pipet by mouth.
f. Stock dithizone solution: See 1070D. 2b1), stock dithizone solution I.
g. Dithizone working solution: Dilute 100 mL stock dithizone solution to 250 mL with CHCl3; 1 mL = 40 μg dithizone.
h. Special dithizone solution: Dissolve 250 mg dithizone in 250 mL CHCl3. This solution may be prepared without purification because all extracts using it are discarded.
i. Sodium sulfite solution: Dissolve 5 g anhydrous Na2SO3 in 100 mL water.
j. Iodine solution: Dissolve 40 g KI in 25 mL water, add 12.7 g resublimed iodine, and dilute to 1000 mL.
a. With sample digestion: To a digested sample containing not more than 1 mL conc acid add 20 mL 1 + 4 HNO3 and filter through lead-free filter paper* and filter funnel directly into a 250-mL separatory funnel. Rinse digestion beaker with 50 mL water and add to filter. Add 50 mL ammoniacal citrate-cyanide solution, mix, and cool to room temperature. Add 10 mL dithizone working solution, shake stoppered funnel vigorously for 30 s, and let layers separate. Insert lead-free cotton in stem of separatory funnel and draw off lower layer. Discard 1 to 2 mL CHCl3 layer, then fill absorption cell. Measure absorbance of extract at 510 nm, using dithizone working solution, ¶ 3g, to zero spectrophotometer.
b. Without sample digestion: To 100 mL acidified sample (pH2) in a 250-mL separatory funnel add 20 mL 1 + 4 HNO3 and 50 mL citrate-cyanide reducing solution; mix. Add 10 mL dithizone working solution and proceed as in ¶ 4a.
c. Calibration curve: Plot concentration of at least five standards and a blank against absorbance. Determine concentration of lead in extract from curve. All concentrations are μg Pb/10 mL final extract.
d. Removal of excess interferences: The dithizonates of bismuth, tin, and thallium differ from lead dithizonate in maximum absorbance. Detect their presence by measuring sample absorbance at 510 nm and at 465 nm. Calculate corrected absorbance of sample at each wavelength by subtracting absorbance of blank at same wavelength. Calculate ratio of corrected absorbance at 510 nm to corrected absorbance at 465 nm. The ratio of corrected absorbances for lead dithizonate is 2.08 and for bismuth dithizonate is 1.07. If the ratio for the sample indicates interference, i.e., is markedly less than 2.08, proceed as follows with a new 100-mL sample: If the sample has not been digested, add 5 mL Na2SO3 solution to reduce iodine preservative. Adjust sample to pH 2.5 using a pH meter and 1+ 4 HNO3 or 1 + 9 NH4OH as required. Transfer sample to 250-mL separatory funnel, extract with a minimum of three 10-mL portions special dithizone solution, or until the CHCl3 layer is distinctly green. Extract with 20-mL portions CHCl3 to remove dithizone (absence of green). Add 20 mL 1 + 4 HNO3, 50 mL citrate-cyanide reducing solution, and 10 mL dithizone working solution. Extract as in ¶ 4a and measure absorbance.
Single-operator precision in recovering 0.0104 mg Pb/L from Mississippi River water was 6.8% relative standard deviation and – 1.4% relative error. At the level of 0.026 mg Pb/L, recovery was made with 4.8% relative standard deviation and 15% relative error.
*Whatman No. 541 or equivalent.
3-70