
In order to promote public education and public safety, equal justice for all, a better informed citizenry, the rule of law, world trade and world peace, this legal document is hereby made available on a noncommercial basis, as it is the right of all humans to know and speak the laws that govern them.
EAS 744:2010
ICS 67.080.01
EAST AFRICAN COMMUNITY
© EAC 2010
First Edition 2010
iDevelopment of the East African Standards has been necessitated by the need for harmonizing requirements governing quality of products and services in East Africa. It is envisaged that through harmonized standardization, trade barriers which are encountered when goods and services are exchanged within the Community will be removed.
In order to meet the above objectives, the EAC Partner States have enacted an East African Standardization, Quality Assurance, Metrology and Test Act, 2006 (EAC SQMT Act, 2006) to make provisions for ensuring standardization, quality assurance, metrology and testing of products produced or originating in a third country and traded in the Community in order to facilitate industrial development and trade as well as helping to protect the health and safety of society and the environment in the Community.
East African Standards are formulated in accordance with the procedures established by the East African Standards Committee. The East African Standards Committee is established under the provisions of Article 4 of the EAC SQMT Act, 2006. The Committee is composed of representatives of the National Standards Bodies in Partner States, together with the representatives from the private sectors and consumer organizations. Draft East African Standards are circulated to stakeholders through the National Standards Bodies in the Partner States. The comments received are discussed and incorporated before finalization of standards, in accordance with the procedures of the Community.
Article 15(1) of the EAC SQMT Act, 2006 provides that “Within six months of the declaration of an East African Standard, the Partner States shall adopt, without deviation from the approved text of the standard, the East African Standard as a national standard and withdraw any existing national standard with similar scope and purpose”.
East African Standards are subject to review, to keep pace with technological advances. Users of the East African Standards are therefore expected to ensure that they always have the latest versions of the standards they are implementing.
© East African Community 2010 — All rights reserved*
East African Community
P O Box 1096
Arusha
Tanzania
Tel: 255 27 2504253/8
Fax: 255-27-2504481/2504255
E-Mail: eac@eachq.org
Web: www.each.int
*© 2010 EAC — All rights of exploitation in any form and by any means reserved worldwide for EAC Partner States’ NSBs.
iiThis standard was developed with support from the Policy Analysis and Advocacy Programme (PAAP) of the Association for Strengthening Agricultural Research in Eastern and Central Africa (ASARECA). This was possible though a grant by the United States Agency for International Development (USAID). This support was used in the process of formulation and mobilization of stakeholders to review the standard in national and regional fora.
ASARECA is a non-political association of agricultural research institutes in: Burundi, DR Congo, Eritrea, Ethiopia, Kenya, Madagascar, Rwanda, Sudan, Tanzania and Uganda. ASARECA serves as a platform for promoting regional research and in the sharing of benefits and spillovers that derive from such research. The mission of ASARECA is to “Enhance regional collective action in agricultural research for development, extension and agricultural training and education, to promote economic growth, fight poverty, eradicate hunger and enhance sustainable use of resources in Eastern and Central Africa”.
Development of standards has been part of PAAP’s contribution to changing the way business is done in crucial agricultural sectors to increase efficiency and/or reduce waste through rationalization and harmonization of policies, laws, regulations and procedures. Rationalization focuses on how countries conduct business in a given subsector, and determines what should be done to make the procedures and processes more efficient. Harmonization brings together regionally different approaches (policies, laws, regulations and procedures) into unified approaches that are applied across the countries. This harmonization process allows commodities and factors to move freely across national boundaries, thereby improving domestic and foreign investment by expanding markets beyond national borders. Over time this will lead to gradual attainment of seamless borders for trade in cassava and cassava products across the region.
Removal of regulatory bottlenecks to transboundary movement of cassava products in the region will enhance competitiveness of trade and value addition in the sub-sector. It will improve the value chains by supporting product differentiation and hence increased trade in cassava products in the region. This will ultimately contribute to incomes, employment generation and improved welfare in the region. This fits snugly with the aspirations of ASARECA as a key player contributing to economic development of the region.
iiiCassava (Manihot esculenta Crantz) is one of the staple food crops. A major drawback of cassava utilization is its potential toxicity due to the presence of endogenous cyanogenic glucosides.
Processing of cassava should reduce the cyanogenic glucosides to an acceptable level. This test method therefore, is intended to be used for determination of the cyanogen content in cassava and its different products as a measure to safeguard the health of the consumers.
ivCassava and cassava products — Determination of total cyanogens — Enzymatic assay method
This East African Standard specifies a method for the determination of total cyanogens in cassava and cassava products.
No normative references have been foreseen for this method.
Linamerase hydrolysis cyanogenic glucoside compounds of cassava extract in orthophosphoric acid medium to produce cyanohydrins which rapidly decompose to cyanide ion in alkali (NaOH), followed by addition of excess pH 6 buffer and chloramine-T to produce a purple solution which is measured spectrophotometrically
Linamarin, stock solution (5 mmol) made by dissolving 31.0 mg of Linamarin in 25 ml buffer, pH 6
NOTE Weighing should be done quickly as the compound is hygroscopic.
Linamarase, stock solution made by dissolving linamerase enzyme in a 0.1 M sodium phosphate buffer to obtain a 2.5 µg linamarin in 0.1 M aliquot
Acetone cyanohydrin (99 %), stock (5 mmol) made by first dissolving 570 μl (0.532 g) in 25 ml of 0.1 M orthophosphoric acid, and from this solution, making 1.00 ml up to 50 ml with 0.1 M orthophosphoric acid.
Because of the poor performance of the pure compound in a pipette, the added quantity should also be weighed for accurate calculation of concentration. The refrigerated stocks can be stored for several weeks to months.
Potassium cyanide (KCN), stock (5 mmol) made by dissolving 163 mg dry and pure KCN in 500 ml 0.2 M NaOH. KCN should be dried for at least 12 h over concentrated H2SO4.
1Standard solutions of 80 μmol and 320 μmol are made from the stock solutions by making 1.60 ml of them up to, respectively 100 ml and 25 ml with 0.1 M orthophosphoric acid. This results in 8 nmol/tube and 32 nmol/tube and absorbance of about 0.220 absorbance unit (AU) and 0.880 AU depending on the spectrophotometer. KCN standards are made just prior to analysis and applied as 0.1 ml to 0.5 ml orthophosphoric acid and 3.4 ml buffer, pH 6 or as 0.1 ml to 3.9 ml buffer, pH 4.
Colour reagent, pure NaOH (3.7 g) is dissolved in 200 ml distilled water. 7 g of 1,3-dimethyl barbituric acid and 5.7 g isonicotinic acid are dissolved in this alkaline solution by extensive stirring. The pH is adjusted between 7 and 8 with 1M HCI or NaOH. This reagent can be kept for at least 12 days at room temperature.
NOTE Stock solutions and reagents should be of analytical grade.
Ordinary laboratory apparatus, and a spectrophotometer
Cut fresh cassava root or moist products into 1 cm cubes and randomly take 50 g cassava cubes and homogenize in 250 ml refrigerated 0.1M orthophosphoric acid in a blender for 15 s at low speed, followed by 60 s at high speed, 60 s of rest and 60 s at full speed again.
For flour 4 g is swirled gently in 25 ml of refrigerated extraction medium in a 50 ml closed container for 5 min.
Extracts are best analyzed immediately, but if not, they should be stored refrigerated or frozen for up to two months.
0.1 ml extract is added to 0.4 ml buffer, pH 7, in a test tube, followed by addition of 0.1 ml linamarase solution. After 15 min incubation at 30 °C, 0.6 ml NaOH (0.2 M) is added, followed after 5 min, by 2.8 ml buffer, pH 6. Coloration is as described in 6.3.
0.1 ml extract is added to 0.6 ml of NaOH (0.2 M). After 2 min, 3.3 ml buffer, pH 6 is added, followed by coloration (6.3).
0.6 ml extract is diluted with 3.4 ml buffer, pH 6 and assayed by colorimetry (6.3).
NOTE Cyanogens are assayed in duplicate and all assays are carried out in glass-stoppered test tubes. The contents of the tubes are mixed after each addition.
20.1 ml Chloramine-T reagent (2 % w/v) is added to the 4 ml buffered extract in the test tubes and mixed. After 5 min, 0.6 ml of color reagent is added and mixed. The absorbance at 605 nm is measured after 10 min. Reagent blanks are run for each analysis. In a digital single beam spectrophotometer, the absorbance can be measured between 0.050 AU and 2.000 AU (Absorbance Unit). In less sophisticated analog equipment, absorbance can be measured between 0.100 AU and 1.200 AU with acceptable accuracy.
Cyanogen levels are calculated in mg HCN equivalent per kilogram sample on dry weight basis (mg HCN equivalent kg-1, DWB) as follows:
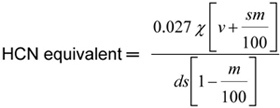
where,
| s | is the sample weight in grams (g); |
| v | is the volume, in millilitres, of the extraction medium; |
| d | is the volume, in millilitres, of extract assayed; and |
| m | is the moisture content, %. |
Dry 5 g to 10 g sample (to constant weight), at 105 °C ± 2 °C (oven) or 70 °C in a vacuum oven and calculate the moisture content (%).
χ is the quantity of cyanogens (nmol) in the tube, calculated from calibration curve as follows:
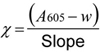
where,
| A605 | is the absorbance measured at 605 nm. Both slope and intercept “w” are derived by linear regression of the calibration points by means of a calculator. For very low values of “w” (<0.005) and higher values of A605 (>0.200) the intercept can be neglected. |
The test report shall show the method used and the result obtained.
It shall also mention any operating conditions not specified in this standard, or regarded as optional, as well as any circumstances that may have influenced the result.
The report shall include all details required for the complete identification of the sample analyzed.
3 4(normative)
Weigh accurately 10 g of the material in a suitable moisture dish previously dried in an electric oven and weighed. Place the dish in an electric oven maintained at 105 °C ± 1 °C for 5 h. Cool the dish in a desiccator and weigh with the lid on. Repeat the process of heating, cooling and weighing at half-hour intervals until the loss in weight between two successive weighings is less than 1 mg.
Record the lowest weight obtained.
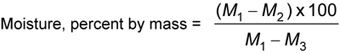
where,
| M1 | is the mass, in grams, of the dish and sample before drying; |
| M2 | is the mass, in grams, of the dish and sample after drying; |
| M3 | is the mass, in grams, of the dish only. |
(normative)
Soxhlet fat extraction apparatus
Petroleum ether, distilling below 65 °C, or ethyl ether
Alcohol potassium hydroxide, 0.1 N (use absolute or alcohol denatured with methanol, [MeOH])
Alcohol-ether mixture, equal volumes of 96 % alcohol and ethyl ether
Phenolphthalein solution, 1 % in alcohol or alcohol denatured with methanol (MeOH) Add 0.3 ml per 100 ml mixture of alcohol-ether and add alcoholic KOH solution to a faint pink.
Extract 10.00 g ± 0.01 g of the sample taken in a thimble with petroleum ether for about 4 h in a Soxhlet extraction apparatus. Completely evaporate the solvent from the extraction flask (weighed previously) on a steam bath, cool and weigh the extraction flask with the residue. Dissolve the residue in the extraction flask with the 50 ml of the alcohol-ether phenolphthalein solution. Titrate the dissolved extract, with standard potassium hydroxide solution, to a faint pink colour, which persists for 10 s. If emulsion is formed during titration, dispel by adding a second 50 ml portion of the alcohol-ether phenolphthalein solution.
Make a blank titration on 50 ml of the alcohol-ether phenolphthalein solution and substract this value from the titration value of the sample. If the additional 50 ml portion of the alcohol-ether phenolphthalein solution is added, double the blank titration.
Calculate the acid value from the following formula:

where,
| V | is the volume, in millilitres, of standard potassium hydroxide solution used; |
| N | is the normality of standard potassium hydroxide solution; and |
| M | is the mass, in grams, of the material taken for the test. |