
In order to promote public education and public safety, equal justice for all, a better informed citizenry, the rule of law, world trade and world peace, this legal document is hereby made available on a noncommercial basis, as it is the right of all humans to know and speak the laws that govern them.
EAS 121:1999
ICS 29:220
EAST AFRICAN COMMUNITY
© EAC 1999
First Edition 1999
iDevelopment of the East African Standards has been necessitated by the need for harmonizing requirements governing quality of products and services in the East African Community. It is envisaged that through harmonized standardization, trade barriers that are encountered when goods and services are exchanged within the Community will be removed.
In order to achieve this objective, the Community established an East African Standards Committee mandated to develop and issue East African Standards.
The Committee is composed of representatives of the National Standards Bodies in Partner States, together with the representatives from the private sectors and consumer organizations. Draft East African Standards are circulated to stakeholders through the National Standards Bodies in the Partner States. The comments received are discussed and incorporated before finalization of standards, in accordance with the procedures of the Community.
East African Standards are subject to review, to keep pace with technological advances. Users of the East African Standards are therefore expected to ensure that they always have the latest versions of the standards they are implementing.
© East African Community 1999 – All rights reserved*
East African Community
P.O. Box 1096
Arusha
Tanzania
Tel: 255 27 2504253/8
Fax: 255 27 2504255
E-mail: eac@eachq.org
Web: www.eachq.org
* © 1999 EAC — All rights of exploitation of any form and by any means reserved worldwide for EAC Partner States’ NSBs
iiIn view of the fact that storage batteries (lead acid type) are now being manufactured in this country a need has been felt to prepare a standard for water for lead-acid-batteries.
This standard gives requirements for distilled or de-ionized water, which should preferably be used whenever it is available and should always be used for counter-EMF cells.
This standard does not cover specifications for water for alkaline cells.
Particular attention should be paid to the storage of water for the maintenance of electric batteries.
Care should be taken to ensure that the container does not contaminate the water; the use of glass or plastic containers is recommended.
In reporting the results of a test or analysis made in accordance with this standard, if the final value, observed or calculated is to be rounded off, it shall be done in accordance with EAS 124:1999, Rounding off numerical values.
In the, preparation of this standard, considerable assistance has been derived from BS 4974, issued by the British Standards Institution.
iii ivWater for lead acid batteries — Specification
This East African Standard specifics requirements for sampling and testing water for lead acid batteries.
For the purpose of examination in accordance with this standard a representative sample of the material not less than 2000 ml in volume shall be taken from the bulk. The sample shall be placed in a clean, dry and airtight glass-stoppered bottle of borosilicate glass of such size that it is nearly filled by the sample. If it is necessary to seal the bottle, care shall be taken to avoid contaminating the contents in any way. For bulk supply, the sampling procedure described in Annex A is recommended.
The material shall be water purified by distillation, ion exchange, or otherwise free from matter in suspension and shall be colourless when viewed through a depth of 300 mm.
The material when tested according to the methods prescribed in Annex B, shall also comply with requirements given in Table 1. Reference to relevant clauses of Annex A is given in column 4 of Table 1.
| SL. No | Characteristics | Requirement | Method of test. (ref to Clause No. Annex B. |
|---|---|---|---|
| 1 | Manganese (as Mn), mg/kg, max. | 0.1 | B.2 |
| 2 | Sulfate residue on ignition, mg/kg, max. | 10 | B.3 |
| 3 | Chloride content (as CI), mg/kg | 10 | B.4 |
| 4 | Ammoniacal nitrogen (as NH), mg/kg, max. | 10 | B.4 |
| 5 | Nitrogen oxides (as N) mg/kg, max. | 10 | B.6 |
| 6 | Iron (as Fe), mg/kg, max. | 10 | B.7 |
| 7 | Copper (as Cu), mg/kg max. | 5 | B.8 |
| 8 | Conductivity in μohms per cm, at 27 °C max. | 1 | B.9 |
The material shall be packed in glass storeware or any other containers as agreed to between the purchaser and the vendor.
The container shall be securely closed and legibly marked with the name of the manufacturer, quality of material in the container, and trademark if any.
The containers shall be marked with identification-code or otherwise to enable the date and lot to be traced back from records.
The containers may also be marked with a Certification Mark.
NOTE The Certification Mark may be used by manufacturers only under licence from EAC. Particulars and conditions under which the licences are granted may be obtained from EAC.
2(normative)
In drawing, preparing, storing and handling samples, the following precautions and directions shall be observed.
Samples shall not be taken in an exposed place.
The sampling instrument shall be clean, before use, these shall be washed several times with the material to be sampled.
Precautions shall be taken to protect the samples, the material being sampled, the sampling instruments and the containers for samples free from adventitious contamination,
To draw a representative sample, the contents of each container selected for sampling shall be mixed as thoroughly as possible by suitable means.
Lot — All containers in a single consignment of the material drawn from a single batch of manufacture shall constitute a lot. If a consignment is declared or known to consist of different batches of manufacture, the batches shall be marked separately and the groups of containers in each batch shall constitute separate lots.
For ascertaining conformity of the material in a lot to the requirements of this standard, samples shall be tested for each lot separately. The number of containers to be selected at random from lots of different sizes shall be in accordance with Table 2.
| Lot size, N |
Sample size, n |
|---|---|
| 3 to 15 18 to 40 41 to 65 66 to 110 111 and above |
3 4 5 7 10 |
In order to ensure randomness of sampling the following procedure shall be adopted. Arrange all containers in a lot in a systematic manner, and starting from anyone, count them as 1, 2 up to r, where
3r is an integral part of 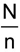 Every r th container thus counted shall be drawn to constitute the test sample n.
Every r th container thus counted shall be drawn to constitute the test sample n.
From each of the containers selected according to A.2.2 equal portions of the material shall be taken out so that the total volume of the quantity collected from all the containers is about 8L. This shall be composite sample.
The composite sample shall be divided into three test samples, not less than 2 L each. These test samples shall be transferred immediately into thoroughly washed bottles which are sealed airtight with glass stoppers and marked with the particulars of sampling, date of sampling, and year of manufacture. One test sample shall be sent to the purchaser and one to the vendor. The third test sample bearing the seals of the purchaser and the vendor shall constitute the referee sample, to be used in case of dispute.
4(normative)
Unless otherwise specified, analytical grade chemicals and distilled water conforming to EAS 123: Distilled water — Specification, shall be used.
The manganese present is oxidized with potassium periodate and the permanganate formed is determined photometrically or alternatively, by visual comparison.
Photoelectric absorptiometer or spectrophotometer with 4 cm cells. Alternatively, matched Nessler cylinders
Eight volumetric flasks, 100-mL capacity
Sulfuric acid, concentrated, 98 % (m/m) (18 mol).
Oxidizing Solution — Mix 250 mL of concentrated nitric acid 70 % (m/m), and 2.5 g of potassium periodate and dilute to 1 000 mL with water.
Standard Manganese Solution — Place in a 400-mL beaker a volume of freshly standardized 0,02 mol potassium permanganate solution calculated from the formula:
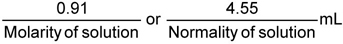
Dilute to about 150 mL and add 5 mL of the sulfuric acid. Carefully add 10 g/L sodium metabisulfate solution, stirring constantly, until the solution is just decolourized. Heat to boil and boil for 10 min. Cool, transfer to a 1 000-mL volumetric flask and dilute to the mark with water. Further dilute 200 ml of this solution to 1 000 mL.
1 ml of the diluted solution = 10 μg Mn.
Preparation of colour standard — Into six 250-mL beakers transfer amounts of the standard manganese solution containing from 0 μg (blank) to 50 μg of manganese, increasing by stages of 10 μg and treat each solution in the manner below.
5Add 16 mL of the sulfuric acid, heat to boil and evaporate until dense white fumes are evolved, reducing the volume to between 5 mL to 10 mL. Allow to cool, cautiously dilute to about 25 mL and 70 mL of, the oxidizing solution. Heat to boil and boil the solution for 4 min, then cool rapidly to room temperature. Transfer the solution to a 100-mL volumetric flask and dilute to the mark with water.
The colour standards are used directly for visual comparison. If an instrument is to be used, measure the absorbances’ of the solution at a wavelength of 520 nm, using the blank as reference standard, and prepare a calibration graph.
Transfer 200 ml of the sample to 500-mL beaker and treat as in B.2.3.2. Measure the absorbance of the solution at a wavelength of 520 nm using the reagent blank as reference standard, and read the amount of manganese present from the calibration graph prepared in B.2.3.3. Alternatively, compare the colour of the solution with the series of prepared colour standards in matched Nessler cylinders, noting the content of the standard that most nearly match the test solution.
The manganese content is expressed as follows:
Manganese content, as milligrams of manganese, Mn, per kilogram = 0.005 m
where
m is the mass of manganese, in μg, in the sample solution found from B.2.3.4
Platinum dish, approximately 50 mm diameter.
Sulfuric acid, 98 % (m/m) (18 mol).
Evaporate carefully, in small portions, 500 mL of the sample to small bulk in a pre-ignited weighed platinum dish. Add a few drops of the concentrated sulfuric acid and evaporate to dryness in a fume cupboard until fumes are evolved. Finally, ignite the residue to constant mass at 850 °C ± 50°C.
Sulfated residue on ignition, in milligrams per kilogram = 2000 × m
where
m is the mass, in g, of the residue found in B.3.3.
The chloride present is determined nephelometrically using silver nitrate.
6Photoelectric absorptiometer or spectrophotometer with 4 cm cells. Alternatively, matched Nessler cylinders.
Fifteen volumetric flasks, 100-mL capacity.
Nitric acid, concentrated 79 % (m/m) (16 mol)
Silver nitrate, approximately 0.1-mol solution
Standard chloride solution — dissolve 1.648 g of previously dried sodium chloride in water and dilute to 1 000 ml. Further dilute 10 mL of this solution to 1 000 ml with water.
1 ml of the diluted solution = 10 μg CI
Preparation of standard solutions — Into thirteen — 100-mL volumetric flasks, each containing about 65 mL of water, transfer amounts of standard chloride solution, containing from 0 (blank) to 120 μg increasing by stages of 10 μg, of chloride, and treat each solution as in B.4.4.2.
Dilute to about 96 ml, add 1 mL of the nitric acid and 2 mL of that silver nitrate solution, dilute to the mark with water, mix well and allow to stand in the dark at room temperature for not less than 6 min. and not more than 16 min.
The solutions are used directly for visual comparison. If an instrument is to be used, measure the absorbance of the solutions in the absorptiometer using a neutral filter and, with the reagent blank as reference standard, prepare a calibration graph.
Determination — Transfer 10 ml of the sample to a 100-mL volumetric flask and dilute to 66 ml with water. Proceed as in B.4.4.2 and measure the absorbance as in B.4.4.3 and read off from the graph prepared In B.4.4.3 the amount of chloride present in the sample. Alternatively, compare the turbidity of the solution with the series of prepared standard solutions in matched Nessler cylinders, noting the chloride content of the standard solution that most nearly matches the test solution.
Chloride content, expressed in milligrams of chlorine, CI, per kilogram = 0.1 × m
where
m is the mass in μg, of the chloride content in the sample found from B.4.4.4.
Ammonia is distilled from the sample after the addition of sodium carbonate and is determined photometrically using Nessler reagent or alternatively by visual comparison.
7Photoelectric absorptiometer or spectrophotometer with 4 cm cell. Alternatively, matched Nessler cylinders.
Twelve volumetric dishes, 100-mL capacity.
All glass distillation apparatus, with 1 000-mL distillation flask.
Distilled water, further purified to free it from ammonia. This can be obtained by shaking 5 000 mL of distilled water with 10 g of strong cation exchange, resin in the hydrogen form or by passing water through a column of such resin. The water may also be prepared by distillation of tap water in all glass apparatus after the addition of 1 mL of a 500-g/l sulfurlc acid solution. The distillate shall be tested with Nessler’s reagent and, when free from ammonia, shall be collected in a glass-stoppered bottle.
Sodium carbonate, anhydrous.
Nessler’s reagent — Dissolve 3.5 g of potassium iodide and 1.25 g of mercury (II) chloride in 80 mL of water. Add a cold saturated aqueous solution of mercury (II) chloride, stirring constantly, until a slight red precipitate remains, then add 12 g of sodium hydroxide. Allow this to dissolve, add little more of the saturated mercury (II) chloride solution until a slight turbidity is obtained, and dilute to 100 mL with water. Allow to settle, decant and, store in the dark.
Standard ammonia solution — Dissolve 3.141 g of ammonium chloride in water and dilute to 1 000 ml. Further dilute 10 mL of this solution to 1 000 mL with water.
1 ml of the diluted solution = 10 μg NH.
Preparation of colour standards — Into eleven of the 10-mL volumetric flasks each containing 60 mL of water, transfer amounts of standard ammonia, solution containing from 0 μg (blank) to 100 μg of ammonia, increasing by stages of 10 μg and proceed as in B.5.4.2,
Dilute to above 95 mL add 2 mL of the Nessler reagent, dilute the mark, mix thoroughly and allow to stand for a period of 10 min to 15 min.
The colour standards are used directly for visual comparison; if an instrument is to be used measure the absorbances of the solution at a wavelength of 370 nm, using the reagent blank as a reference standard and prepare a calibration graph.
Determination Transfer 60 mL of the sample to the distillation flask to 500 mL with water and add 0.5 g of sodium carbonate. Connect the flask to the distillation assembly heat to boil and collect 100 mL of distillate. Transfer a 20 mL aliquot portion to another 1 000 mL volumetric flask and proceed as in B.5.4.3. At the same time, carry out a blank test using the reagents alone. Measure the absorbance of this solution as in B.5.4.4 using the blank as reference standard and read the amount of ammonia present from the calibration graph obtained in B.5.4.3. Alternatively compare the colour of the solution with the series prepared colour standards in matched Nessler cylinders, noting the amount of ammonia content of the standard that most nearly matches the test solution.
Collect a further 100 mL distillate and treat in the same way; no ammonia should be present in this second portion of distillate.
8Ammoniacal nitrogen content expressed milligrams of ammonia, NH, per kilogram = 0.1 m
where
m is the mass of ammonia, in μg found in the aliquot portion of the sample taken.
The nitrogen oxides present are first oxidized using potassium permanganate solution and then treated with 2, 4-xylenol reagent. The resulting nltro-xylenol is distilled and the amount present in the distillate is determined photometrically or alternatively by visual comparison.
Photoelectric absorptiometer with 4 cm cell. Alternatively, matched Nessler cylinders
Water bath, capable of being controlled at 35 °C ± 1°C
Twelve volumetric flasks, 100 mL capacity
All-glass distillation apparatus with 250 mL distillation flask
Mercury (II) Acetate
Sulfuric acid, 15.5-mol solution. Add 400 ml of concentrated sulfuric acid 98 % (m/m) (18 mol) (free of nitrogen) to 100 mL of water. To ensure complete elimination of nitrogen oxides, prepare the concentrated sulfuric acid as follows: Cautiously add 450 mL of concentrated sulfuric acid to approximately 110 mL of water and heat until white fumes are liberated. Cool, repeat the dilution and heating twice.
Hydrogen peroxide 1 g/L solution
2.4 xylenol, 10 g/L solution in glacial acetic acid, freshly prepared.
Sodium hydroxide, approximately 2-mol solution.
Sodium hydroxide, approximately 0.2-mol solution. Dilute 100 ml of the 2-mol sodium hydroxide solution (see B.6.3.5) to 100 mL; with water.
Potassium permanganate 0.02 mol solution
Standard nitrogen solution, dissolve 0.360 9 g of potassium nitrate in water and dilute to 1 000 mL. One millilitre of the solution = 500 μg N
9Preparation of standard distillate — To 9 mL of water contained in a small flask, carefully add 30 mL of the sulfuric acid solution. Cool to 36 °C, then pour the diluted acid into a 100 mL ground glass stoppered boiling tube containing 1.00 mL of the standard nitrogen solution.
Add 1 mL of the 2,4-xylenol reagent and mix well. Transfer the tube to the water bath and maintain at 35 °C ± 1 °C for 30 min. At the end of this period, remove from the bath and transfer the contents of the tube completely to the distillation flask and dilute with 100 mL of water. Connect the flask to the distillation flask and dilute with 100 mL of water. Connect the flask to the distillation assembly heat to boiling and collect approximately 60 ml of the distillate in a 100-mL volumetric flask containing 10 mL of the 2 mol sodium hydroxide solution. Cut the water supply to the condenser towards the end of the distillation so as to ensure that no nitrate 2,4-xylenol remains in the condenser. Dilute to the mark with water and mix thoroughly.
Preparation of colour standards — Into ten of the 100 mL volumetric flasks, transfer amounts of from 1 mL to 10 mL of the distillate, increasing in stages of one millilitre corresponding to nitrogen content from 5 μg to 50 μg. Dilute each solution to the mark with the 0.2 mol sodium hydroxide and mix thoroughly.
These colour standards are used directly for visual comparison. If an instrument is used, measure the absorbances of the solution at a wavelength 445 nm, using the 0.2 mol sodium hydroxide as a reference standard, and prepare a calibration graph.
Determination — Transfer 10 mL of the sample to a 100 mL ground glass stoppered boiling tube. Add 2 to 3 drops of the potassium permanganate solution and 0.5 mL of the sulfuric acid solution drop by drop, from a burette, shaking the tube during the addition. Then add hydrogen peroxide solution, drop by drop, until the permanganate colour is just discharged. Immerse the tube in ice water and, when cold, add a further 29.5 mL of the sulfuric acid solution drop by drop, mixing continuously, keeping the temperature below 40 °C during this addition. Add 0.2 g of the mercury (II) acetate and when dissolved, proceed as B.6.4.2. Carry out a blank test on 30 ml of the sulfuric acid solution adding it drop by drop to 10 mL water contained in a 100-mL ground glass stoppered boiling tube in ice water, keeping the temperature below 40 °C during the addition. Add 0.2 g of mercury (II) acetate and when dissolved, proceed as B.6.4.2.
Measure the absorbance of the blank solution at a wavelength of 445 mm, using the 0.2 mol sodium hydroxide solution as reference standard. Subtract the absorbance of the blank from that of the test solution and read the amount of nitrogen present from the calibration graph. Alternatively compare the colour of the test and blank solutions with the series of prepared colour standards in matched Nessler, cylinders. Note the nitrogen contents of the matching standards and determine the nitrogen content of the sample by difference.
Nitrogen, expressed as milligrams of Nitrogen, N, per kilogram = 0.1 m.
where
m is the mass of nitrogen in μg as found from the calibration graph.
The iron present is reduced to iron (II) state and determined photometrically using 2,2’-bipyridyl or alternatively by visual comparison.
10Photoelectric absorptiometer or spectrophotometer with 4 cm cells. Alternatively, matched Nessler cylinders
Thirteen volumetric flasks 100-ml capacity
One volumetric flask, 250 ml capacity
Hydrochloric acid, approximately 1 mol solution
Hydroxyl ammonium chloride, 50 g/L solution
Ammonium acetate, 400 g/L solution
2.2’-bipyridyl 1 g/L solution — Dissolve 0.1 g of the reagent in 50 ml of water containing 2 ml of the 1 mol hydrochloric acid solution and dilute to 100 mL.
Standard iron solution — Dissolve 7.022 g of ammonium Iron (II) sulfate in a mixture of 600 mL of water and 360 ml of concentrated sulfuric acid (98 % m/m) (18 mol) and dilute to 1 000 mL with water. Further dilute 10 mL of the solution so obtained to 1 000 mL with water.
1 ml of the diluted solution = 10 μg Fe
Preparation of colour standards — Into eleven of the 100 mL volumetric flasks, each containing from 50 mL of water and 2 mL of the 1 mol hydrochloric acid solution, transfer amounts of the standard iron solution, containing from 0 μg (blank) to 100 μg of iron, increasing by stages of 10 μg and treat each solution as in B.7.4.2
Add 4 mL of the hydroxyl ammonium chloride solution and allow standing for 60 s. Add 6 ml of ammonium acetate solution and 3 ml of the 2.2’-bipyridyl solution. Dilute the content of each flask with water to 100 mL and mix thoroughly.
These colour standards are used directly for visual comparison. If an instrument is to be used, measure the absorbances of the solution, at a wavelength of 515 nm, using the reagent blank as a reference standard and prepare a calibration graph,
Determination — Transfer 10 mL of the sample into a 100 mL volumetric flask. Add 2 mL of the 1-mol hydrochloric acid solution, 4 mL of the hydroxyl ammonium chloride solution, and allow to stand for 60 s. Add 5 mL of the ammonium acetate solution, mix and add 3 mL of the 2.2’-bipyridyl solution. Dilute to 100 mL with water and mix thoroughly. At the same time carry out a blank test on the reagents alone.
Measure the absorbance of the solution at a wavelength of 515 nm, using the reagent blank as reference standard, and read the amount of iron present from the calibration graph.
Alternatively compare the colour of the solution with the series of prepared colour standards in matched Nessler cylinders noting the iron content of the standard that most nearly matches the test solution.
11Iron content, expressed in milligrams of Iron, Fe, per kilogram = 0.1 m
where
m is the mass of iron, in μg, as found from the calibration graph.
The copper present is reduced with ascorbic acid and a violet coloured complex is formed by addition of 2.2’-biquinolyl. This complex is extracted with amyl alcohol and measured photometrically or alternatively by visual comparison.
Photoelectric absorptiometer or spectrophotometer with 4 cm cells. Alternatively, matched Nessler cylinders
Eight volumetric flasks, 50-mL capacity
Sodium sulfate, anhydrous
Hydrochloric acid, 1 mol solution
Amyl alcohol
Hydrochloric acid, concentrated, 36 % (m/m) (11 mol)
Tartaric acid, 500 g/L aqueous solution
Sodium hydroxide, 5 mol aqueous solution
Ascorbic acid, 100 g/L aqueous solution, freshly prepared.
2.2’-biquinolyl, 0.5 g/L solution in amyl alcohol.
Standard copper solution — Dissolve 0.392 8 g of copper (II) sulfate pentahydrate in water, add 26 mL of 3 mol sulfuric acid solution, and dilute to 1 000 mL. Further dilute 10 mL of the solution to 100 mL.
1 mL of the diluted solution = 10 μg Cu.
Narrow range indicator papers, covering the range pH 5.5 to 7.0.
Preparation of colour standards — Into six 100 mL beakers transfer amounts of standard copper solution containing from 0 μg (blank) to 100 μg of colour, increasing by stages of 20 μg and treat each solution as in B.8.4.2.
12Dilute to approximately 30 mL with water and transfer to a 600 mL stoppered separating funnel. Dilute with water to approximately 400 ml and add 2 mL of the tartaric acid solution. Adjust the pH of the solution to approximately 6.0 by addition of sodium hydroxide solution using a narrow range indicator paper externally. Add 2 mL of the ascorbic acid solution, shake to mix thoroughly and allow to stand for 5 min. Add 10 mL of the 2,2’-biquinolyl solution and shake well for about 2 min. Extract the copper complex with two 20 mL portions of amyl alcohol and transfer the extract to 100 mL beaker. Add about 2 g of the anhydrous sodium sulfate to the combined extracts and stir thoroughly to remove traces of water. Fitter into a 60 mL volumetric flask end wash the sodium sulfate crystals twice with 2 mL portions, of amyl alcohol Transfer the washings to the flask and dilute to the mark with amyl alcohol.
The colour standards are used directly for visual comparison. If an instrument is to be used, measure the absorbances of the solutions at a wavelength of 45 nm, using the reagent blank as reference standard, and draw a calibration graph.
Determination — Transfer 10 mL of the sample into a 500 mL stopperred-separating funnel. Add 1 mL of the concentrated hydrochloric acid, and proceed as from the second sentence of B.8.4.2. At the same time carry out a blank test on the reagents alone. Measure the absorbance of the solution at a wavelength of 545 nm, using the reagent blanks solution reference standard and read the amount of copper present in the sample from the calibration graph in B.8.4.3. Alternatively, compare the colour of the solution with the series of prepared colour standards in matched Nessler cylinders, noting the copper content of the standard that most nearly matches the test solution.
Copper content, expressed as milligram of copper, Cu, per kilogram = 0.1 m
where
m is the mass of copper, in μg, in sample found from the calibration graph.
A sample of the water is boiled in a conical flask for a period of 5 min. The flask is sealed and the flask and contents cooled to 27 °C ± 2 °C and conductivity measured within 15 min.
Conical flask, 500 mL capacity, lined with ground glass joint
Guard tube, fitted with a ground glass joint, containing soda (self-indicating) nominal aperture size 0.71 mm to 1.18 mm
Conductivity meter
Transfer 400 mL of the water into the flask. Heat until the water boils and continue boiling for 5 min. Remove the flask from the heat and immediately insert the guard tube. Cool the flask end contents rapidly under running water. Measure the conductivity within 15 min cooling to 27 °C ± 2 °C.
13 14